Introduction
promoris a user-friendly, comprehensive R package that combines proteomics data analysis with machine learning-based modeling.promorstreamlines differential expression analysis of label-free quantification (LFQ) proteomics data and building predictive models with top protein candidates.promorprovides a range of quality control and visualization tools at the protein level to analyze label-free proteomics data.Input files for
promorare a proteinGroups.txt file produced by MaxQuant or a standard input file containing a quantitative matrix of protein intensities and an expDesign.txt file containing the experimental design of your proteomics data.The standard input file should be a tab-delimited text file. Proteins or protein groups should be indicated by rows and samples by columns. Protein names should be listed in the first column and you may use a column name of your choice for the first column. The remaining sample column names should match the sample names indicated by the mq_label column in the expDesign.txt file.
Installation
You can install the development version of promor from GitHub with:
# install devtools, if you haven't already:
install.packages("devtools")
# install promor from github
devtools::install_github("caranathunge/promor")Proteomics data analysis with promor
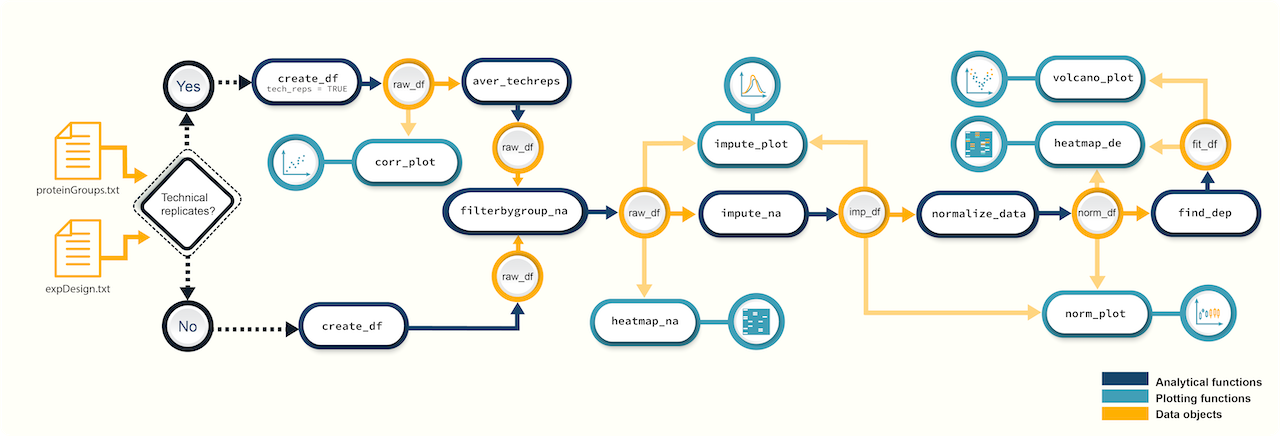
Figure 1. A schematic diagram of suggested workflows for proteomics data analysis with promor.
Example
Here is a minimal working example showing how to identify
differentially expressed proteins between two conditions using
promor in five simple steps.
We use a previously published data set from Cox et al. (2014) (PRIDE ID: PXD000279).
# Load promor
library(promor)
# Create a raw_df object with the files provided in this github account.
raw <- create_df(
prot_groups = "https://raw.githubusercontent.com/caranathunge/promor_example_data/main/pg1.txt",
exp_design = "https://raw.githubusercontent.com/caranathunge/promor_example_data/main/ed1.txt"
)
# Filter out proteins with high levels of missing data in either condition/group
raw_filtered <- filterbygroup_na(raw)
# Impute missing data and create an imp_df object.
imp_df <- impute_na(raw_filtered)
# Normalize data and create a norm_df object
norm_df <- normalize_data(imp_df)
# Perform differential expression analysis and create a fit_df object
fit_df <- find_dep(norm_df)Lets take a look at the results using a volcano plot.
volcano_plot(fit_df, text_size = 5)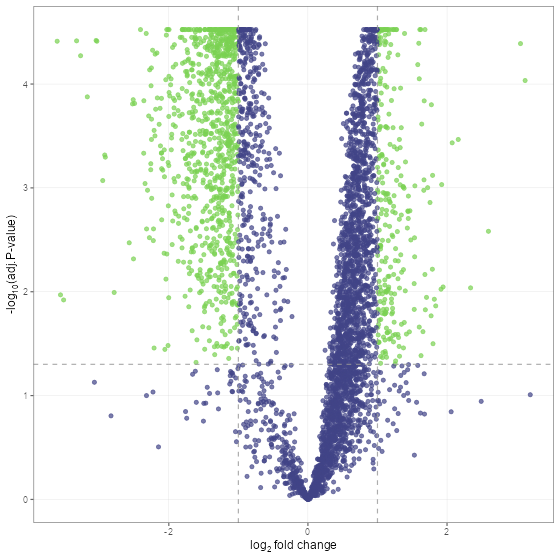
Modeling with promor
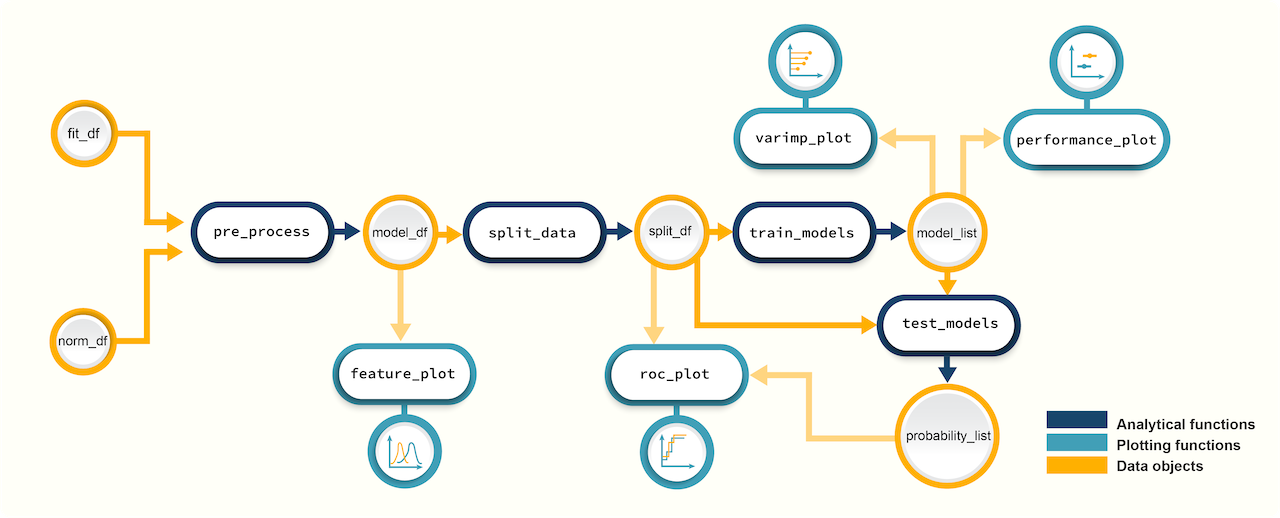
Figure 2. A schematic diagram of suggested workflows for building predictive models with promor.
Example
The following minimal working example shows you how to use your results from differential expression analysis to build machine learning-based predictive models using promor.
We use a previously published data set from Suvarna et al. (2021) that used differentially expressed proteins between severe and non-severe COVID patients to build models to predict COVID severity.
# First, let's make a model_df object of top differentially expressed proteins.
# We will be using example fit_df and norm_df objects provided with the package.
covid_model_df <- pre_process(
fit_df = covid_fit_df,
norm_df = covid_norm_df
)
# Next, we split the data into training and test data sets
covid_split_df <- split_data(model_df = covid_model_df)
# Let's train our models using the default list of machine learning algorithms
covid_model_list <- train_models(split_df = covid_split_df)
# We can now use our models to predict the test data
covid_prob_list <- test_models(
model_list = covid_model_list,
split_df = covid_split_df
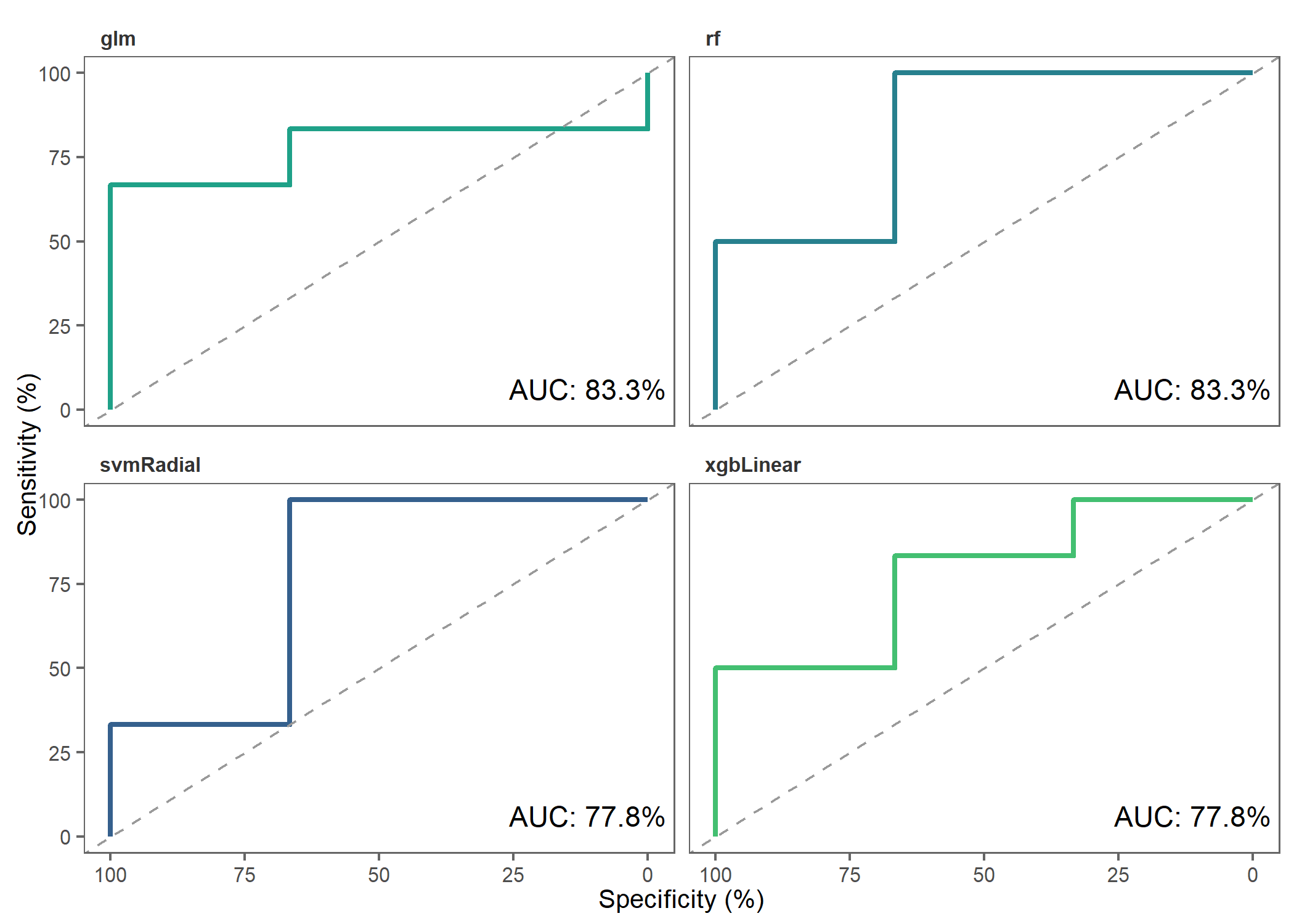
)Let’s make ROC plots to check how the different models performed.
roc_plot(
probability_list = covid_prob_list,
split_df = covid_split_df
)
Tutorials
You can choose a tutorial from the list below that best fits your experiment and the structure of your proteomics data.
If your data do NOT contain technical replicates: promor: No technical replicates
If your data contains technical replicates: promor: Technical replicates
If you would like to use your proteomics data to build predictive models: promor: Modeling